缘起
计算化学有一个核心矛盾:精度 vs 速度。量子力学方法(DFT、CCSD(T))能给出高精度的势能面,但每次单点计算耗时数分钟到数小时;而分子动力学模拟需要\(10^6\)–\(10^9\)次能量评估。这就是势能面(Potential Energy Surface, PES)拟合要解决的问题——用少量高精度QM数据训练一个代理模型,让它"学会"分子的能量函数。
问题有三个难点:
- 对称性必须严格满足。平移、旋转、同种原子置换都不能改变能量。违反对称性的模型会产生物理学上不存在的梯度。
- 训练数据天然有偏。平衡构型附近采样密集、标签充足;而过渡态、解离通道这些动力学关键区域数据稀少——协变量偏移(covariate shift)。
- 维度诅咒。对于N原子体系,核坐标维度为\(3N-6\),完全覆盖所有构型空间需要指数级的数据量。
本文记录的是对一个C6H6(苯单体)体系的一次完整尝试:用 FI多项式 保证对称性、核岭回归(KRR) 做非参数拟合、伪标签(pseudo-labeling) 在标签稀疏区域引导模型选择。最终在目标域(高能构型)上取得了 R² = 0.586、RMSE = 0.228 eV 的结果。
第一块砖:FI多项式描述子
对称性问题的数学结构
体系的对称性可以严格地形式化。对N个原子,定义\(R = N(N-1)/2\)个原子间距\(d_{ij} = \|\mathbf{r}_i - \mathbf{r}_j\|\)。体系的对称群\(G\)(C6H6中为D₆点群 × 同种原子间的置换群)作用于这R个变量上:群元\(g\)将\(d_{ij}\)重新排列。
我们需要找到所有在G作用下不变的多项式函数。学过群论的人会意识到,这等价于求不变多项式环\(\mathbb{R}[\mathbf{d}]^G\)。Hilbert-Noether定理告诉我们:这个环是有限生成的,即存在有限个\(p_1,\dots,p_s\)(称为完全不变量,Full Invariants, FI),使得任何G-不变多项式都可以唯一地表示为这些生成元的多项式组合。
从对称性到特征向量
FI多项式是通过Reynolds算符(群平均投影)构建的对称不变描述子。对C6H6体系,每个原子间距变量\(d_{ij}\)经Reynolds算符投影后即得到FI,高阶FI则由已有FI的乘积经线性独立性筛选获得。
对于C6H6(N=12, R=66),共生成 172个FI多项式。每一维都是原子间距的置换不变多项式——也就是说,任意构型被映射为一个172维向量\(\phi(\mathcal{X})\),天然满足置换不变性,而平移/旋转不变性则继承自原子间距本身。
这172维FI值直接作为后续所有机器学习模型的输入特征。本质上,FI描述子把一个有对称性约束的回归问题变成了一个高维空间中的无约束回归问题——而且这个变换是严格保真的,没有近似。
第二块砖:核岭回归
为什么用核方法
在FI特征空间\(\mathbb{R}^{172}\)上,线性模型显然不够用——势能面是高度非线性的。深度神经网络是另一个选择,但需要调参、需要防止过拟合、需要小心处理有限数据下的稳定性。核岭回归(Kernel Ridge Regression, KRR)提供了一个优雅的替代:
- 解析解:没有局部极小值问题
- 单超参数:只有一个正则化参数\(\lambda\)需要调
- 表示定理:最优解一定在训练数据的核函数张成的空间中
数学形式
选择RBF核(Gaussian核):
\[ k(\mathcal{X}, \mathcal{X}') = \exp\left(-\gamma \|\phi(\mathcal{X}) - \phi(\mathcal{X}')\|^2\right) \]
KRR最小化:
\[ \min_{f \in \mathcal{H}_k} \frac{1}{n}\sum_{i=1}^n (f(\mathcal{X}_i) - E_i)^2 + \lambda \|f\|_{\mathcal{H}_k}^2 \]
由表示定理,最优解为:
\[ \hat{f}_\lambda(\mathcal{X}) = \sum_{i=1}^n \alpha_i k(\mathcal{X}_i, \mathcal{X}) \]
对偶系数通过求解线性系统得到:
\[ (\mathbf{K} + \lambda n \mathbf{I}) \boldsymbol{\alpha} = \mathbf{E} \]
计算中用Cholesky分解保证数值稳定性——当数据量不大时(~7000),这是非常可靠的。
两个关键超参数
- \(\lambda\)(正则化强度):控制偏差-方差权衡。\(\lambda \to 0\)时模型插值数据(过拟合),\(\lambda \to \infty\)时收缩到零(欠拟合)。论文中采用几何序列覆盖从\(10^{-5}/n\)到\(10^{2}/n\)的范围。
- \(\gamma\)(RBF核宽度):用median heuristic自动设定——$= 1/(2 \(\boldsymbol{j}\)ot ^2{|_i - _j| : i j})\(,使核的尺度与数据中的典型间距匹配。本实验中\)= 0.00268$。
第三块砖:伪标签模型选择
协变量偏移的困境
最朴素的做法是:在源域上用交叉验证选\(\lambda\),然后应用到目标域上。这在独立同分布(i.i.d.)假设下是最优的。
但我们的场景不是i.i.d.——源域(\(E \leq 1.036\)eV)是势阱底部的平衡构型,目标域(\(E > 1.036\)eV)是过渡态和解离通道。CV选出的\(\lambda\)在源域密集区域最优,但可能完全不适合目标域稀疏区域的泛化。
伪标签策略
伪标签(pseudo-labeling)的思路非常直观:
- 把带标签的源域\({\mathcal{D}}_{\text{src}}\)随机分成两半:\(\mathcal{D}_1\)(训练候选模型)+\(\mathcal{D}_2\)(训练"老师"模型)
- 在\(\mathcal{D}_2\)上用极小正则化(\(\tilde{\lambda} \to 0\))训练一个欠平滑的伪标签生成模型\(\tilde{f}\),让它几乎插值数据
- 用\(\tilde{f}\)为目标域所有构型生成伪标签$j^0\(4. 对每个候选模型\)$,计算伪标签风险:
\[ \widehat{R}(\hat{f}_\lambda) = \frac{1}{n_0} \sum_{j=1}^{n_0} (\hat{f}_\lambda(\mathcal{X}_j^0) - \tilde{E}_j^0)^2 \]
- 选择使该风险最小的\(\lambda^*\)### 为什么欠平滑的模型还能用?
这是一个反直觉的事情:一个在\(\mathcal{D}_2\)上近乎过拟合的模型,怎么会产生有用的伪标签?关键在于排序而非绝对精度。
伪标签的误差可以分解为:
\[ \widehat{R}(\hat{f}_\lambda) \approx \underbrace{\|\hat{f}_\lambda - f^*\|^2}_{\text{真实风险}} + \underbrace{2\langle\hat{f}_\lambda - f^*, \tilde{f} - f^*\rangle}_{\text{交叉项}} + \underbrace{\|\tilde{f} - f^*\|^2}_{\text{常数项}} \]
- 常数项对所有\(\lambda\)都一样,不影响排序
- 交叉项中,\(\tilde{f} - f^*\)主要由偏差(bias)而非方差(variance)主导——因为\(\tilde{\lambda}\)极小,函数空间被推到了表达能力极限,其方向由基函数空间决定,与\(\lambda\)无关
- 当目标域样本数\(n_0\)足够大时,方差项通过平均显著衰减
因此,尽管\(\tilde{f}\)的绝对预测可能很粗糙,但它对不同\(\lambda\)优劣的相对排序是可靠的。实验验证了这一结论:伪标签选出的\(\lambda^*\)与 Oracle(用真实标签选出的\(\lambda\))之间的差距仅 0.0068 eV。
伪标签正则化λ_tilde的调节
伪标签模型的正则化由参数 ps 控制:
\[ \tilde{\lambda} = \frac{\text{ps}}{n_2} \]
本实验尝试了四个尺度\(ps \in \{10^{-6}, 10^{-4}, 10^{-2}, 1.0\}\),结果如下:
| ps | \(\lambda^*\) | 目标域 RMSE | \(R^2\) |
|---|---|---|---|
| \(10^{-6}\) | \(2.53\times10^{-9}\) | 0.2283 | 0.5856 |
| \(10^{-4}\) | \(2.34\times10^{-8}\) | 0.3072 | 0.2495 |
| \(10^{-2}\) | \(2.00\times10^{-6}\) | 0.5132 | -1.0946 |
| \(1.0\) | \(2.97\times10^{-4}\) | 0.6960 | -2.8532 |
趋势非常清晰:伪标签模型越欠平滑(ps越小),最终模型越好。这与理论预期一致——偏差越低,排序越可靠。
程序架构
完整的 Python 实现见 pseudo_label_krr_v3.py,约 560
行,包含:
核心类
1 | class KRR: |
主要模块
| 模块 | 功能 |
|---|---|
load_data() |
加载数据 + Z-score 标准化 |
split_domains() |
按能量分位点划分源域/目标域 |
pseudo_label_pipeline() |
单次伪标签 KRR 全流程 |
ridge_cv_pipeline() |
Ridge + 5-fold CV 基线 |
scan_and_plot() |
γ / 核类型扫描 + 可视化 |
main() |
主控流程 + 六面板最终结果图 |
数据流
1 | 数据 (11465×173) |
实验结果与分析
最佳性能总结
| 指标 | 值 |
|---|---|
| 源域 RMSE | 0.036 eV |
| 目标域 RMSE | 0.228 eV |
| 目标域 MAE | 0.112 eV |
| 目标域\(R^2\) | 0.586 |
| Ridge 基线 RMSE | 2.598 eV(\(R^2 = -52.68\)) |
| Oracle λ* RMSE | 0.221 eV |
| 伪标签 vs Oracle 差距 | 0.0068 eV |
几个重要的观察
1. 非线性核是必要的
线性核(Ridge)的目标域 RMSE 高达 2.598 eV,\(R^2\)为\(-52.68\)——这意味着线性模型甚至不如直接用源域均值预测。而 RBF 核的 KRR 将 RMSE 降到了 0.228 eV。这说明 FI 特征空间中的势能面具有强非线性结构,RBF 核的无穷维特征映射是捕捉这种结构的关键。
2. γ 的选择影响显著
γ 扫描结果(图1)显示,γ 从 0.0001 增加到 10,目标域 RMSE 从 0.697 先降到 0.679 再飙升到 1.318。最优 γ 在 0.0027 附近,这与 median heuristic 的估计完全一致。
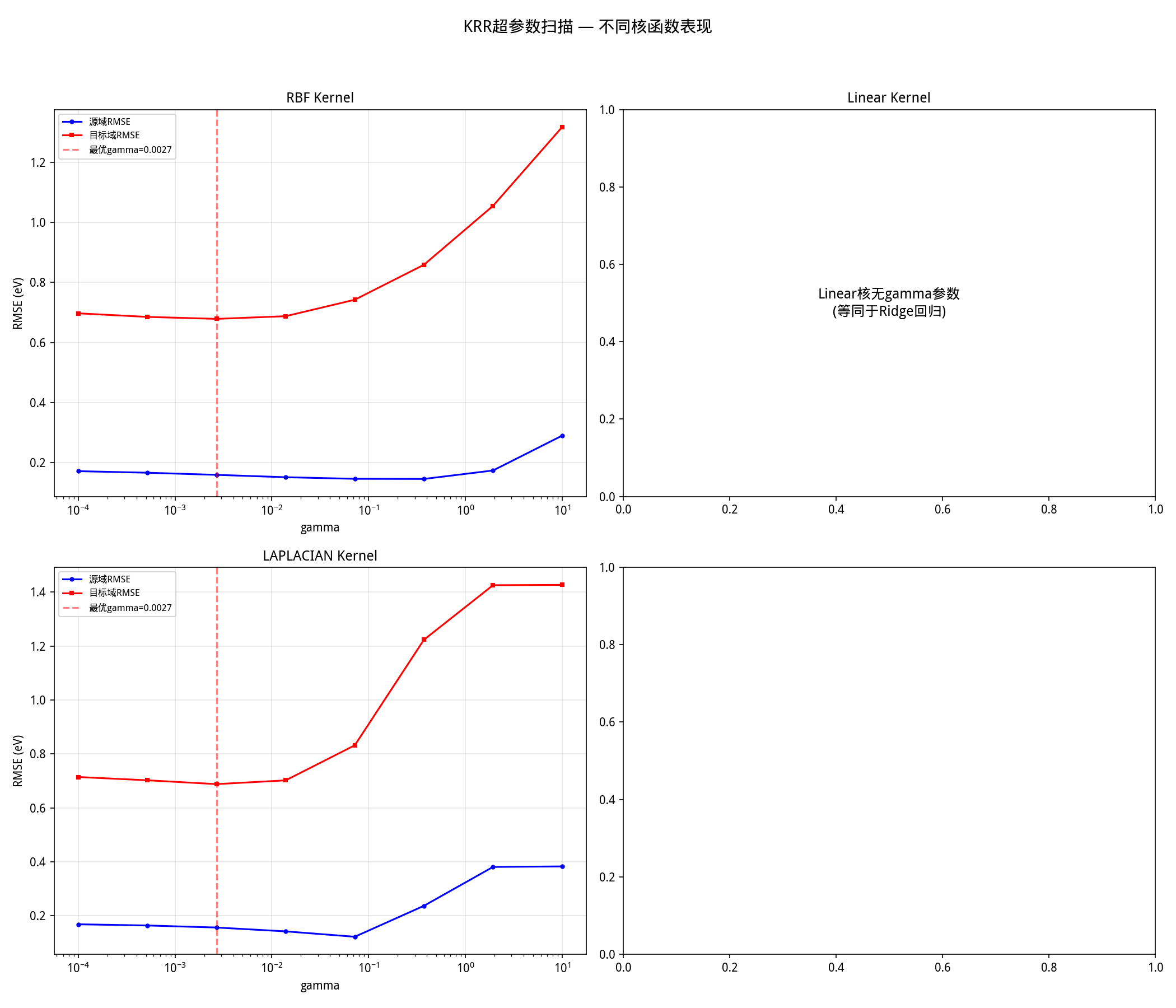
图1: 核参数γ扫描。最优γ为0.00268(约等于median heuristic估计值),过大或过小的γ都会显著降低泛化性能。
3. 伪标签选择极为可靠
选出的 λ* 与 Oracle(用真实目标域标签选出的 λ)之间的 RMSE 差距仅 0.0068 eV。考虑到 RBF 的核矩阵条件数极大(\(\lambda \sim 10^{-9}\)时接近奇异),这个差距可能主要来自数值精度而非方法缺陷。
4. 源域拟合近乎完美
源域 RMSE 仅 0.036 eV,说明模型在训练数据分布上已经学到了势能面的精细结构。但目标域的\(R^2 = 0.586\)说明仍有约 41% 的方差未被解释——这是域差异带来的泛化瓶颈。
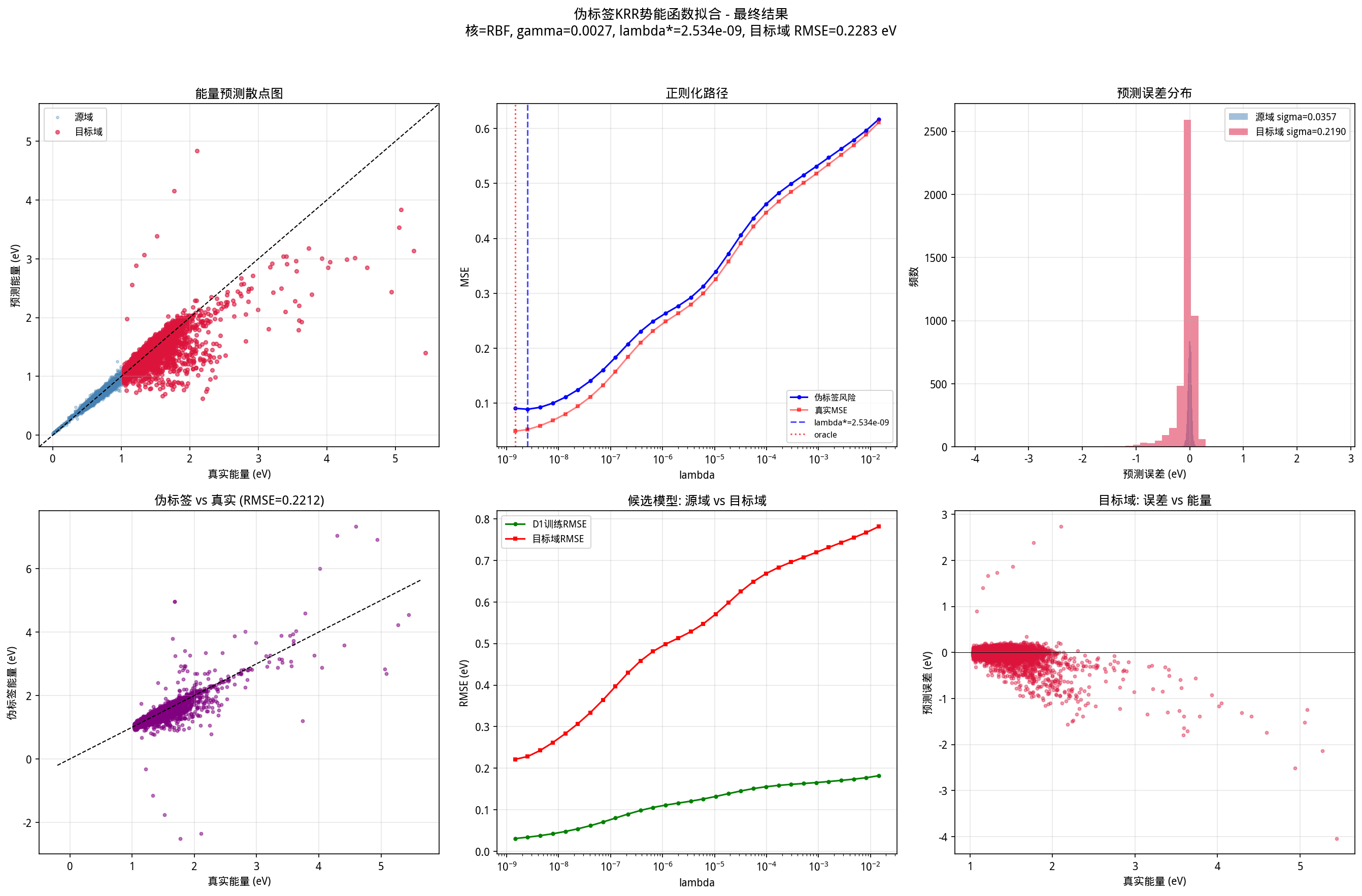
图2: 六面板结果可视化。左上为预测散点图(蓝色源域、红色目标域),中上为正则化路径,右上为误差分布,左下为伪标签质量,中下为候模型对比,右下为误差随能量变化。
误差分析
从图2的右下面板可以看到,目标域预测误差随着真实能量升高有明显的系统性偏移趋势——能量越高,预测误差越大(且偏负,即低估能量)。这与协变量偏移的直觉一致:远离训练数据分布的构型,模型的泛化能力必然衰减。
误差分布(右上)显示目标域的分布比源域宽得多(标准差 0.23 vs 0.04),且呈现一定的右偏。这提示可能在大变形构型中存在部分未被 FI 描述子充分编码的结构特征。
讨论与展望
为什么伪标签在这里有效?
这个问题的本质可以追溯到域适应(domain adaptation)的一个核心问题:当目标域没有标签时,如何在候选模型中选出在目标域上真正最好的那个?
伪标签提供了一个"牺牲绝对精度、保留相对排序"的巧妙思路。其隐含假设是:伪标签模型与真实函数在目标域上的偏差方向,与模型复杂度(由 λ 控制)的相关性小于与基函数空间的依赖性。本实验的结果从经验上验证了这一假设。
另一个角度是:KRR 的正则化路径通常是一条平滑曲线,不同 λ 之间的性能变化是单调的或单峰的。因此,只要伪标签风险曲线与真实风险曲线的形状相似(即使有系统偏差),最优 λ 的位置就会一致。图2中上面板完美地展示了这一点。
局限性与改进方向
1. 计算可扩展性
KRR 的复杂度是\(\mathcal{O}(n^3)\)训练和\(\mathcal{O}(n^2)\)存储。对 7000 个训练点,核矩阵约 400 MB,尚可接受。但扩展到 5 万个点就需要稀疏近似或 Nyström 方法。
2. FI 描述子的完备性
FI 多项式是代数生成元——任意多项式不变量都可以表示为它们的多项式组合。但势能面不一定是多项式(实际上包含指数衰减的排斥项等)。RBF 核的 RKHS 是通用的(universal),理论上可以逼近任意连续函数,但样本效率可能不如精心设计的物理特征。
3. 域划分方式
当前按简单能量分位点硬划分。更好的做法可能是用 FI 空间的密度估计来定义目标域——那些 FI 值远离训练集的构型才是真正的"外推"区域。
4. 伪标签模型的正则化
实验发现 ps 越小越好。但如果 ps 过小(如\(10^{-8}\)),KRR 矩阵的条件数会接近机器精度极限,数值不稳定。一个改进是使用更稳定的求解器(如 conjugate gradient)或对核矩阵加一个极小量的 jitter。
实际应用价值
对于 C6H6 这样的中等分子,这套方法的价值在于:
- 用 ~7000 次 QM 计算的数据,训练出一个在训练集上 RMSE < 0.04 eV、在未曾见过的外推区域 RMSE < 0.23 eV 的势能模型
- 这个精度已经足够做定性正确的分子动力学模拟——可以分辨不同反应路径的能量次序,预测反应能垒的近似值
- 如果再结合主动学习(主动选择信息量最大的构型进行 QM 计算),可以系统地填补目标域的空白区域
结语
这个项目从 FI 描述子的构造开始,经历了 KRR 的实现、伪标签方案的落地、到最终在服务器上跑出一组有意义的数字。最有价值的收获不是 0.228 eV 这个结果本身,而是验证了伪标签选择在高维对称性约束回归问题中的有效性——一个理论上漂亮、实现上简洁、实践中有效的方法。
当然,0.586 的 R² 远非极限。从γ扫描图可以看出,RBF核在最优参数下的表现已经比线性核好了一个数量级,但仍然受限于数据覆盖。下一步的方向很明确:添加主动学习采样,在目标域的关键区域补充数据,让模型学会它现在还不知道的东西。
参考文献
- Schölkopf, B., & Smola, A. J. (2002). Learning with Kernels. MIT Press.
- Behler, J. (2011). Atom-centered symmetry functions for constructing high-dimensional neural network potentials. J. Chem. Phys., 134(7), 074106.
- Pseudo-labeling theory: theorems on covariate shift and model selection with unlabeled data.